
Miaomiao Zhang, Yuting Gao, Lanya Fu, Yimin Bai, Somnath Mukherjee, Cheng-Lung Chen, Jing Liu*, Hongtao Bian*, and Yu Fang. J. Phys. Chem. Lett., 2023, 14, 6968-6976.
In the field of lithium-ion batteries (LIBs), aqueous electrolytes have garnered extensive attention due to their superior safety and ionic conductivity. These aqueous electrolytes exhibit long cycling life and significant energy density, making them highly promising for large-scale applications in energy storage systems and devices. However, the narrow electrochemical stability window of water has been a persistent challenge in this field. The emergence of "Water-in-salt" (WIS) electrolytes offers a potential solution by expanding the electrochemical stability window of water, thus significantly advancing the development of aqueous electrolytes. However, in highly concentrated aqueous solutions, the conduction of Li+ and their local solvation structure remain not fully understood. Understanding the solvation structure of Li+ and specific ion-ion interactions is crucial for understanding their migration behavior and determining macroscopic viscosity and conductivity. Ion diffusion in solution generally occurs through two modes: vehicular migration and hopping transport. Vehicular migration involves the diffusion of ions and their tightly bound solvated layer as a whole, while hopping transport is a process of ion diffusion through successive dissociation/association exchanges between solvation layers. The mechanism of Li+ ion conduction in aqueous electrolyte solutions is currently unclear. Some previous studies suggest that rearrangement of hydrogen bonding networks plays a role in achieving vehicular migration. However, due to the rapid timescale (faster than 1 nanosecond) of hydrogen bonding network rearrangement, a consensus on the conduction mechanism of Li+ in aqueous electrolytes has not been reached.
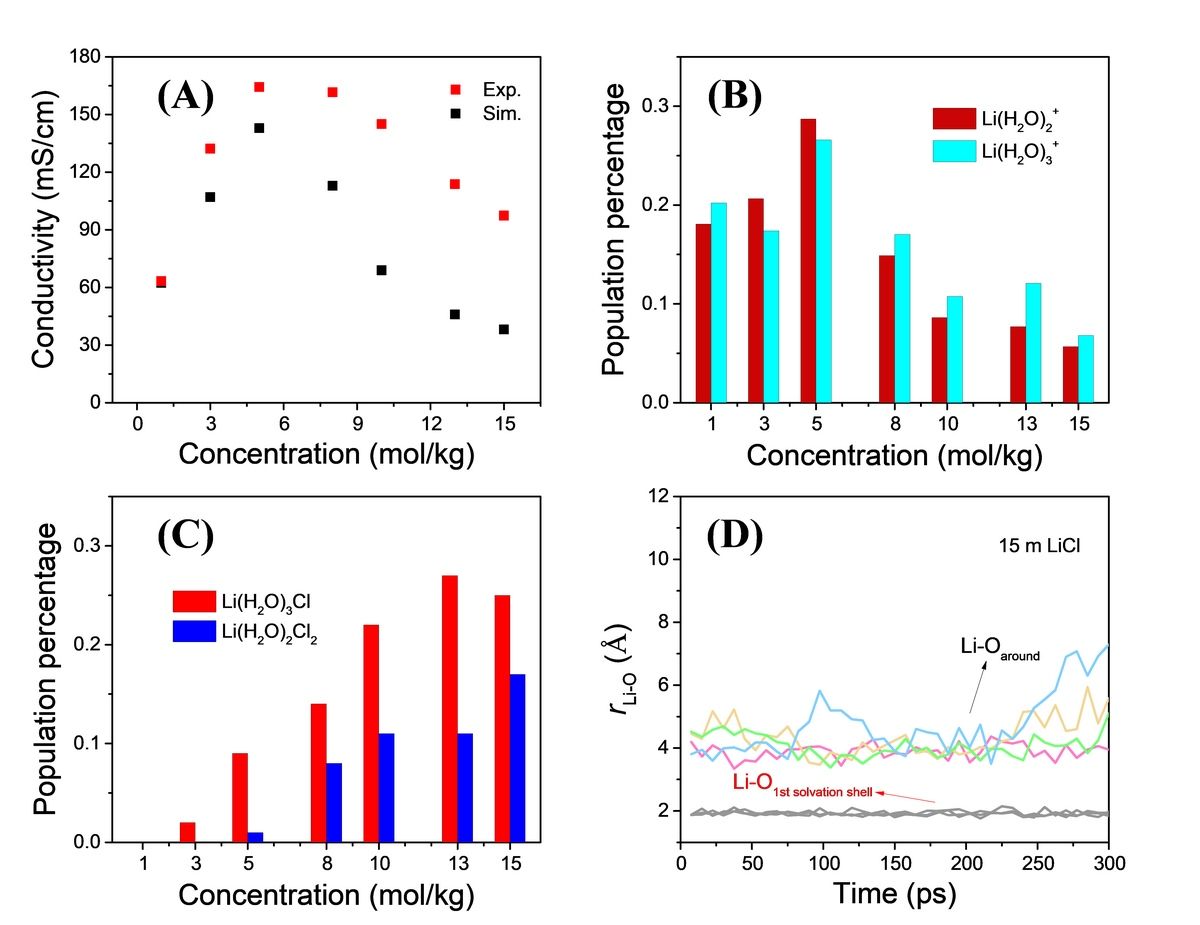
To solve this issue, we used ultrafast infrared spectroscopy and molecular dynamics simulations to investigate the structure and dynamics of Li+ in highly concentrated LiCl water solutions. Ultrafast infrared spectroscopy and thiocyanate (SCN) were used as local anionic vibrational probes to study the structural dynamics of Li+ in LiCl water solutions. The experimental results indicated that the infrared frequencies and vibrational relaxation dynamics of the external probe strongly depend on the bulk concentration, effectively probing the local environment and structural dynamics of Li+, while overcoming the limitations of using OD stretching vibrations as probes in previous literature. By using the Stokes-Einstein-Debye (SED) equation, the coupling between the reorientational dynamics of the external probe in LiCl water solutions and the macroscopic viscosity was analyzed, revealing the formation and structural dynamics of ion-water complexes. To obtain the spatial structural heterogeneity of LiCl water solutions, molecular dynamics (MD) simulations were performed. These simulations revealed the formation of chain-like structures consisting of chloride ions and Li+ through electrostatic interactions at high concentrations. These extended chain-like structures strongly constrained the reorientation of water molecules around them, impacting the dynamics and transport of Li+. Through MD simulations, the concentration-dependent conductivity of LiCl water solutions, which is in good agreement with experimental results, was obtained. Further analysis revealed that Li(H2O)2+ and Li(H2O)3+ were the main ion species contributing to Li+ ion conduction. The study also performed a detailed analysis of selected lithium ions and oxygen atoms in water molecules (r(Li-O)) in 15 m LiCl solutions over a time range of 300 ps. The trajectory analysis supported the vehicular migration of Li+ through the dynamic chain-like structure formed between solvents and ions. The Cartesian coordinates analysis of Li+ indicated that the r(Li-O) distance between Li+ and water molecules in the first solvation shell remained constant at about 2.0 Å, indicating stable complex formation with the adjacent three waters. However, the surrounding water molecules moved closer and then further away within a given time range but never penetrated the solvated shell to enter the inner solvation sheath, suggesting that Li+ ion diffusion mainly occurs through vehicular transport rather than hopping. The experimental and MD simulation results indicate that Li(H2O)3Cl and Li(H2O)2Cl2 are the major determinants of the viscosity of LiCl solutions. These specific Ion-water complexes and chain-like structures facilitate the vehicular migration of Li+ through the aqueous electrolytes, providing a deeper understanding of the origin of viscosity and conductivity in highly concentrated electrolyte aqueous solutions.
This study provides important insights into the structural dynamics and transport mechanism of Li+ in highly concentrated aqueous electrolytes. Understanding the role of chain-like structures and ion-water complexes in Li+ ion conduction may guide the development of novel aqueous electrolytes for use in lithium-ion batteries. Establishing correlations between ion pair structures and macroscopic properties can also facilitate the design of safer and more efficient lithium-ion batteries, promoting the widespread application of aqueous electrolytes in the field of energy storage.
First Author: Zhang Miaomiao, Doctoral candidate, Shaanxi Normal University
Corresponding Authors: Prof. Bian Hongtao, Prof. Liu Jing, Shaanxi Normal University
Full Text Link: https://pubs.acs.org/doi/10.1021/acs.jpclett.3c01494







